For downloading myPresto newest version. myPresto is a program suite composed of several molecular simulations for drug development.


Table of Contents
Simple software package with graphic interface
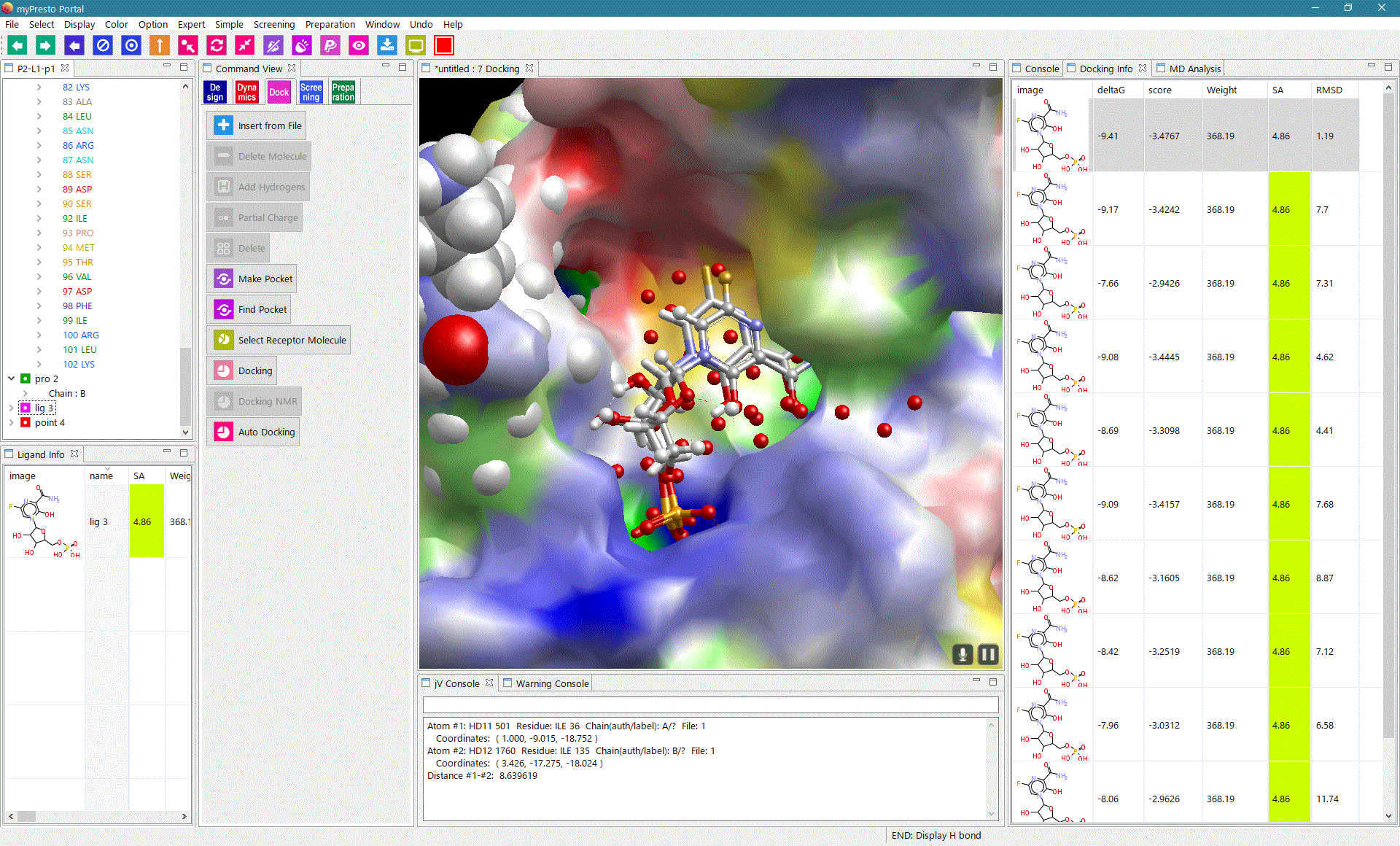
myPresto Portal
myPresto Protal is a graphic user interface (GUI) program for myPresto and GROMACS, available for Windows, MAC and Linux (all 64bit). |
Easy myPresto
Easy myPresto is a graphic user interface (GUI) program for myPresto, available for Windows, MAC and Linux (all 64bit). |
| The free version is distributed through the generosity of member companies, with restrictions on molecular weight, functions, etc., to paid software. | |
| MolDesk EC shop | Paid version of Easy myPresto |
Molecular dynamics simulation
cosgene: COnformation SamplinG ENginE
A molecular dynamics simulation program for protein, DNA, chemical compound, protein-ligand complex. AMBER and CHARMM force fields are available. Cosgene can calculate NVE, NVT and NPT ensemble with various boundary conditions. In addition, cosgene can calculate the generalized ensemble like multicanonical ensemble. Cosgene can calculate the protein-compound binding free energy by the filling potential method. The generalized ensemble method is useful to calculate the free energy surface.
Fukunishi, Y., Mikami, Y., Nakamura, H. (2003) J. Phys. Chem. B. 107, 13201-13210
Last update on February 23, 2020
psygene-G: Protein dYnamics SimulatinG ENginE for GPU
A rapid molecular dynamics simulation program, which uses our original non-Ewald algorithm (Zero-Dipole summation method ) for electrostatic interactions, has been developed for a single and multiple GPUs (Graphics Processing Units) with the space decomposition algorithm.
Mashimo, T., Fukunishi, Y., Kamiya, N., Takano, Y., Fukuda, I., Nakamura, H. (2013) J. Chem. Theory Comput. 9, 5599-5609
Last update on November 7, 2019
omegagene: a GPU-accelerated molecular dynamics simulator with enhanced conformational sampling
A new engine, “omegagene”, has joined in myPresto program suits for more effective conformational sampling with GPU-accelerated molecular dynamics simulation. It incorporates our original non-Ewald algorithm, Zero-Multipole summation Method (ZMM), and an improved Multicanonical molecular dynamics (v-McMD) algorithm. This program has been developed mainly at Osaka University, and it is also distributed under Open-Source License from http://www.protein.osaka-u.ac.jp/rcsfp/pi/omegagene/.
Kasahara, K., Ma, B., Goto, K., Dasgupta, B., Higo, J., Fukuda, I., Mashimo, T., Akiyama, Y., Nakamura, H. (2016) Biophys. & Physicobiol. 13, 209-216: DOI 10.2142/biophysico.13.0_209
Last update on January 14, 2018
PDBcheck
Checks the validity of input PDB files, determines N- and C-terminal modifications and disulfide bonds (S-S) of proteins, and edits H-amino acid states of metal-binding sites.
setwater
Add solvent water molecules to systems containing proteins, DNA/RNA, membranes, and small molecules in spherical (isolated) and rectangular (periodic) forms.
add_ion
Neutralizes the charge with NaCl at a specified concentration and adjusts the salt concentration to an arbitrary level for isolated and periodic systems hydrated with setwater containing proteins, DNA/RNA, membranes, and small molecules.
Gromacs compatible tools
convertTpl、ctl2mdp.pl、log2form.pl
Creation of systems containing proteins, nucleic acids, and small molecules is easy with myPresto. myPresto converts the system files created with myPresto into input for Gromacs, a fast MD software. Gromacs output files can be converted into highly readable myPresto format files.
Structure preparation and file format conversion for small molecules, etc.
Hgene: Hydrogen Generation ENginE
Addition and deletion of hydrogen atoms in dissociated and nondissociated forms at around pH 7 for small and medium organic molecules, and simple 3D structure generation. Add or remove hydrogen atoms in dissociated and non-dissociated forms, generate simple 3D structures, and add Gasteiger and MOPAC AM1BCC charges to low and medium organic molecules, MOPAC PM3 for structure optimization.
Supported formats are SMILES / ISIS MOL, SDF / Sybyl mol2 / PDB / mmCIF
Last update on March 23, 2022
Force-field parameter assignment
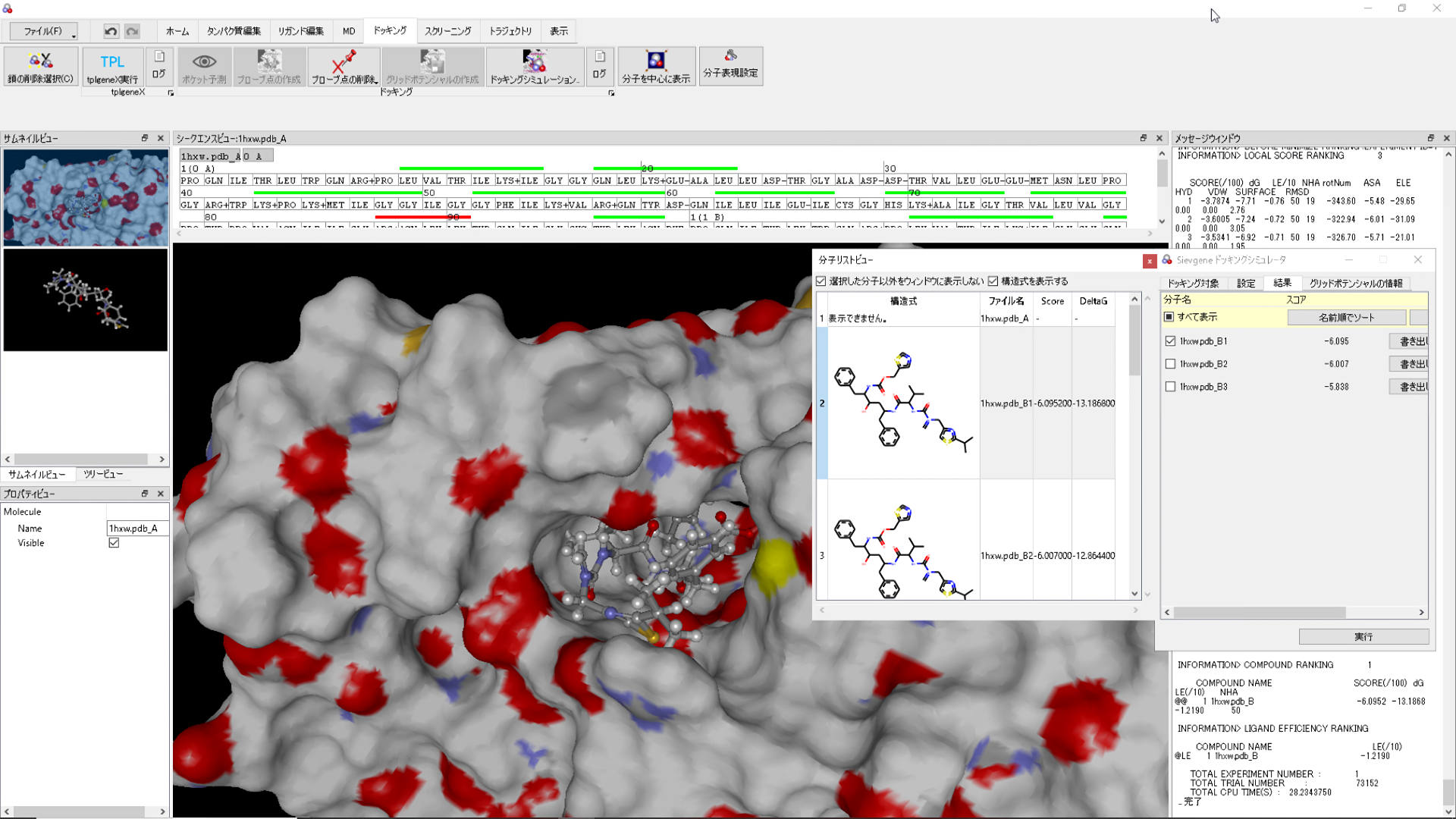
tplgeneX: ToPoLogy generatinG ENginE eXtended
This engine is used to construct molecular topologies of biological macromolecules for use in other myPresto programs.
It newly allows input/output of atomic coordinates in PDBx/mmCIF format and simplified handling of ligand molecule topologies.
Supported molecules: Proteins (including phosphorylated and methylated), nucleic acids (RNA / DNA), membrane molecules, water, various salts, typical solvents (DMSO, monosaccharides, etc.), metals, etc.
Supported force field:
AMBER force fields: ff99, ff99-Fuji, ff99-SB, ff99-SB-ILDN, ff99-SB-DISP
Nucleic acid force field: ff99, ff99bsc0, OL3, YIL, ROC, Shaw
Membrane molecular force field: lipid14
Last update on March, 2022
tplgeneL: ToPoLogy generatinG ENginE for Ligands
This engine is for generating molecular topology of a target ligand (chemical compound) for usage in other programs in myPresto.
Last update on January 12, 2018
Membrane system
membgene3
A tool program for modeling a system including transmembrane protein, lipid bilayer membrane, water, and ions.
Last updated on October 2, 2021
Free energy calculation
Filling potential method to calculate a binding free energy between two molecules
In this method, free energy profiles are obtained from the analysis of many MD trajectories which are obtained with MD calculations with umbrella potentials.
Last updated on March 11, 2018
Conformer generation
Confgene:Conformer Generation ENginE
(included in ToolsYYMMDD.tar.gz)
A conformer generator for compound. Confgene can generate various conformers of compound.
Last update on January 18, 2018
ConfgeneC: Conformer Generation ENginE for Cyclic part
(included in ToolsYYMMDD.tar.gz)
A conformer generator for cyclic part of compound.
Last update on January 18, 2018
descgene
Generate molecular descriptor files by randomly generating conformations including ring conformations and linear chains of small molecules.
(radius of inertia, ASA, flatness, eccentricity, MACCS key/Joback descriptor, etc.)
Compound database
3DdataConstruction
A tool box for 3D mol2 file generation from 2D mol file. It can generate 3D compound database from a 2D chemical compound catalog.
Last update on December 3, 2019
LigandBox: LIGANd Data Base Open and eXtensible
This is an open database, containing the three-dimensional (3D) molecular structures and atomic charges of chemical compounds, which are available from industries. Some are collected from the KEGG DRUG database.
If you want LigandBox, please specify “Your Affiliation” and apply with Contact. We will inform you of the download site of LigandBox by email.
Sample of molecule files : Mol2 SDF
{kind=link}
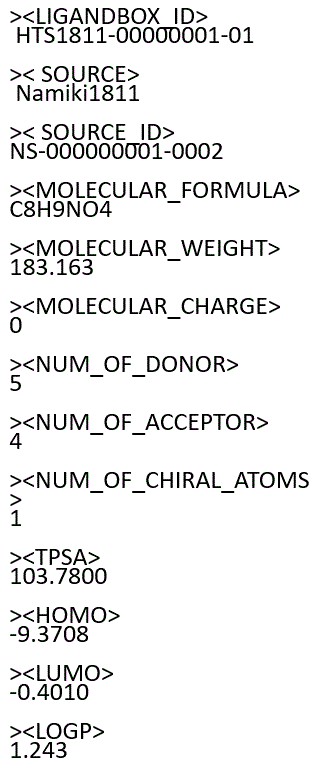{kind=link}
Receptor-ligand binding site search
MolSite
A ligand-binding site prediction. MolSite predicts ligand-binding site of target protein and its affinity by a compound docking simulation.
Y. Fukunishi, H. Nakamura. “Prediction of ligand-binding sites of proteins by molecular docking calculation for a random ligand library.” Protein Science, in press.
Last update on January 12, 2018
sitegene
Search for concave areas on the molecular surface of proteins and nucleic acids.
UAP: Univeral Active Probes
The universal active probe (UAP) is a set of drug-like compounds. This method can select the reliable drug-screening result among many screening results, since the hit ratio of the UAP is proportional to the hit ratio of the true active compounds.
Y. Fukunishi, K. Ono, M. Orita, H. Nakamura. “Selection of in-silico drug screening result by using universal active probes (UAPs).” Journal of Chemical Information and Modeling, 2010, 50, 1233-1240
Last update on January 19, 2018
Protein-compound docking
sievgene: SIEVinG ENginE
A protein-compound docking program for in-silico (virtual) drug screening and prediction of protein-ligand complex structure. Sievgene is a fast flexible docking program. On average, it can dock one compound to a target in 1 seconds with high accuracy (56% complex structures satisfy RMSD < 2A in a self-docking test) on usual PC. It is designed for PC grid computing.
Fukunishi, Y., Mikami, Y., Nakamura, H. (2005) J. Mol. Graph. Model. 24, 34-45
Last update on March 20, 2018
sievgene_M
sievgene_M is a modified version of sievgene, which can be also executed on a system with Intel Xeon Phi. Inte Xeon Phi is a new CPU including more than 50 cores.
Last update on January 20, 2018
sievgeneDIAV: SIEVinG ENginE with DIAV method
A protein-compound docking program, sievgene, with DIAV method to estimate protein-compound binding free energy more correctly than the original sievgene does (less than about 1.3 kcal/mol).
Fukunishi, Y., Nakamura, H. (2013) Pharmaceuticals 6, 604-622
Last update on May 7, 2022
sievgeneNMR: SIEVinG ENginE for NMR
A protein-compound docking program for prediction of protein-ligand complex structure with using NMR signal of ligand. The prediction accuracy is improved approximately twice comparing to the original sievgene by using the DIRECTION NMR signal of ligand.
Fukunishi, Y., Mizukoshi, Y., Takeuchi, K., Shimada, I., Takahashi, H., Nakamura, H. (2012) J. Mol. Graph. Model. 31, 20-27
Last update on August 24, 2019
sievgeneMVO: SIEVinG ENginE for Maximum Volume Overlap
A protein-compound docking program for prediction of protein-ligand complex structure / drug screening with using a given known-ligand docking pose. This software performs a ligand docking and overlapping the ligand onto the given known-ligand structure given by the user at the same time. It is a combination of docking and similarity search.
Fukunishi, Y., Nakamura, H. (2012) Pharmaceuticals 5, 1332-1345
Last update on January 11, 2018
Docking-score QSAR
Docking-score QSAR method is to predict protein−compound binding energies from known activity.
Fukunishi, Y., Yamashita, Y., Mashimo, T., Nakamura, H. (2018) Molecular Informatics 37, 1868-1743
Last update on February 16, 2020
in-silico drug screening
selectMTS: Multiple Target Screening method
(included in screening_packYYMMDD.tar.gz)
A structure-based drug screening program based on protein-compound affinity matrix. selectMTS can perform the multiple target screening (MTS) method, machine-learning MTS (MSM-MTS) method, direct score modification MTS (DSM-MTS) method. If active compounds of your target are known, a machine-learning MTS can show high hit ratio.
Y. Fukunishi, Y. Mikami, S. Kubota, H. Nakamura, “Multiple target screening method for robust and accurate in silico screening.” Journal of Molecular Graphics and Modelling,25, 61-70 (2005)
Y. Fukunishi, S. Kubota, H. Nakamura, “Noise reduction method for molecular interaction energy: application to in silico drug screening and in silico target protein screening”, Journal of Chemical Information and Modeling, 46, 2071-2084 (2006)
Last update on October 29, 2018
selectDSI: Docking Score Index method
(included in screening_packYYMMDD.tar.gz)
A ligand-based drug screening program based on protein-compound affinity matrix. selectDSI can perform the docking score index (DSI) method and machine-learning DSI (ML-DSI) method. If active compounds of your target are known, a machine-learning DSI can show high hit ratio.
Y. Fukunishi, Y. Mikami, K. Takedomi, M. Yamanouchi, H. Shima, H. Nakamura. “Classification of chemical compounds by protein-compound docking for use in designing a focused library”, Journal of Medicinal Chemistry, 49, 523-533 (2006)
Y. Fukunishi, S. Hojo, H.Nakamura, “An efficient in silico screening method based on the protein-compound affinity matrix and its application to the design of a focused library for cytochrome P450 (CYP) ligands.”, Journal of chemical information and modeling, 46, 2610-22 (2006)
Last update on January 12, 2018
Affinity_fingerprint: Compound database and protein-compound affinity matrix
Our 3D chemical compound database consists of more than 10 millions compounds. Also, we prepared several sets of protein-compound affinity matrix generated by sievgene. The matrix is a matrix of docking scores of 182 proteins vs. 2 millions compounds. This database is essential to use selectMTS and selectDSI. Please contact Yoshifumi Fukunishi, phD (y-fukunishi * aist. go. jp: is the e-mail address. please replace * by @, and remove the space “ “.) to get these databases. The size of these databases is beyond the limit of FTP download, so that we distribute these databases by hard disk drive.
Substrucure_search
A ligand-based drug screening program based on chemical compound structure comparison. A substructure search method based on the molecular structure by using the Ullman’s method.
Last update on January 12, 2018
TGS: Topological Graph Search (A Similarity Search Using Molecular Topological Graphs)
A similarity search method based on eigen values of molecular edge matrix.
Y. Fukunishi, H. Nakamura. “A similarity search using molecular topological graphs”, Journal of Biomedicine and Biotechnology, Volume 2009 (2009), Article ID 231780
Last update on January 13, 2018
MVO: Molecular-Dynamics Maximum-Volume Overlap
(included in ToolsYYMMDD.tar.gz)
A similarity search method based on overlap of molecular volume by molecular dynamics simulation.
Y. Fukunishi, H. Nakamura, “A new method for in-silico drug screening and similarity search using molecular-dynamics maximum-volume overlap (MD-MVO) method”, Journal of Molecular Graphics and Modelling, 2009, 27, 628-636
Last update on January 18, 2018
MVO_screening
A ligand search program using the Moleclar volume overlap (MVO) method.
Last updated on February 27, 2018
Physical property estimation
Ligand property prediction
This is a tool for predicting physical properties of low molecular weight compounds and medium molecules using regression learning. Using learned data, predicted values of membrane permeability coefficient (LogPa, apparent permeability), distribution coefficient considering ionization (LogD, distribution coefficiene), and solubility (LogS) can be calculated.
Y. Fukunishi, T. Mashimo, T. Kurosawa, Y. Wakabayashi, H.K. Nakamura, K. Takeuchi. “Prediction of Passive Membrane Permeability by Semi‐Empirical Method Considering Viscous and Inertial Resistances and Different Rates of Conformational Change and Diffusion.” Molecular informatics, 2019:38, 1900071
Last update on January 30, 2022
LogS_prediction
A solubility (LogS) predictor based on molecular descriptor and salvation free energy calculation. Aggregators (frequent hitters) can be predicted based on the calculated LogS values.
T. Mashimo, Y. Fukunishi, M. Orita, N. Katayama, S. Fujita, H. Nakamura. “Quantitative analysis of aggregation-solubility relationship by in-silico solubility prediction.” International Journal of High Throughput Screening, 2010:1, 99-107
Last update on January 14, 2018
Synthetic accessibility estimation and Drug design
ReCGen: Generate virtual compounds based on input structure
This is the source code for a fragment-based virtual compound generator program.
The program offers the 3 following tools: Fragment database creator tool, database merge tool, structure generator tool.
The copyright for ReCGen, a compound structure generator program, is owned by Kyoto Constella Technologies Co., Ltd.
Last updated on March 10, 2018
SyntheticAccessibility: Prediction of the synthetic accessibility of given compounds.
This tool predicts the synthetic accessibility (SA) of given compounds based on the complexity, number of chiral centers and symmetry of the compound. The SA is shown as real number from 0 (easy) to 10 (difficult).
Y. Fukunishi, T. Kurosawa, Y. Mikami, H. Nakamura. Prediction of Synthetic Accessibility Based on Commercially Available Compound Databases. Journal of Chemical Information and Modeling. (2014), 54 (12), 3259-3267.
Last updated on January 11, 2018
Interface for new PDB format
LibMyPresto: Package of the library programs for parsing and writing PDBx/mmCIF and PDB formatted files.
Library programs to parse and write either PDBx/mmCIF or PDB formatted files, with the users programs in C, f77 and f90 languages.
Last updated on January 19, 2018